


.png)

.png)


.png)

Open source AI model matches the performance of AlphaFold3
Dec. 20, 2024.
2 mins. read.
 4 Interactions
4 Interactions
MIT scientists have released a new open source AI model to predict protein structure with a performance similar to AlphaFold3.
About the Writer





MIT scientists have released Boltz-1, a new artificial intelligence (AI) tool that could have a significant impact on how we do biomedical research and make new drugs.
Boltz-1 is a fully open source model that matches the performance of AlphaFold3, a model from Google DeepMind that guesses the shapes of proteins and other biological molecules.
Proteins are vital for all biological functions, and their shape decides what they do. Knowing a protein’s shape is key to making new drugs or designing protein functions.
The scientists have presented Boltz-1 at an MIT event, hoping it would encourage worldwide teamwork and speed up scientific discoveries. They want Boltz-1 to be a starting point for further community work.
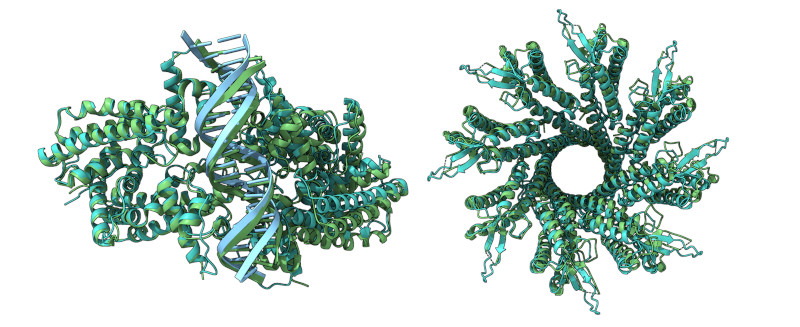
Predicting protein shapes is tough because of the complex way amino acids fold into 3D structures. AlphaFold2, which won a Nobel Prize, used machine learning to predict these shapes very accurately. AlphaFold3 added a new AI method called a diffusion model, which helps deal with the uncertainty in these predictions. But AlphaFold3 isn’t fully open-source, leading to some pushback and a race to make an open-source version.
Democratizing research in biology
The Boltz-1 code is publicly available via GitHub. The Boltz-1 GitHub repository links to a technical reports titled “Boltz-1 Democratizing Biomolecular Interaction Modeling,” published in bioRxiv.
Boltz-1 started with the same methods as AlphaFold3 but added improvements to make it more accurate. They also shared all the code for training Boltz-1, making it easier for others to improve or use it. This project took four months, and one big challenge was handling the Protein Data Bank’s vast and varied data.
Boltz-1 matches AlphaFold3’s accuracy in predicting various biomolecular structures. The team plans to keep enhancing Boltz-1, reducing prediction times, and they invite others to join in via GitHub or Slack. Experts see Boltz-1 as a breakthrough, democratizing access to advanced tools in biology, which could lead to new medicines, and predict a surge in discoveries thanks to this open-source model.
Let us know your thoughts! Sign up for a Mindplex account now, join our Telegram, or follow us on Twitter.








0 Comments
0 thoughts on “Open source AI model matches the performance of AlphaFold3”